iMeta | 上海交大张晨虹组-解析膳食纤维干预期间的微生物群动态
点击蓝字 关注我们人体肠道微生物群对膳食纤维的时间响应模式iMeta主页:http://www.imeta.science研究论文● 原文: iMeta (IF 23.8)● 原文链接: https://onlinelibrary.wiley.com/doi/10.1002/imt2.70046● DOI: https://doi.org/10.1002/imt2.70046● 2025年5月10
点击蓝字 关注我们
人体肠道微生物群对膳食纤维的时间响应模式

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接: https://onlinelibrary.wiley.com/doi/10.1002/imt2.70046
● DOI: https://doi.org/10.1002/imt2.70046
● 2025年5月10日,上海交通大学张晨虹等在iMeta在线发表了题为“Temporal response patterns of human gut microbiota to dietary fiber”的文章。
● 本研究对19名超重受试者(部分伴有2型糖尿病)在14天正常饮食及随后14天膳食纤维干预期间,进行了每日血糖、肠道微生物组成及代谢物的动态监测。通过基于个体网络的功能群分析和时间序列分析,捕捉了肠道菌群的动态响应,揭示了在传统采样策略下常被忽视的多种菌群丰度变化模式。结合多组学数据和时滞效应分析,进一步识别出与宿主代谢改善相关的关键微生物和代谢产物,为后续机制研究提供了可靠靶点。
● 第一作者:林晓彤、王朝迅
● 通讯作者:张晨虹(zhangchenhong@sjtu.edu.cn)
● 合作作者:刘标、朱寅、翟芮
● 主要单位:上海交通大学生命科学技术学院、微生物代谢全国重点实验室,上海浦东医院内分泌科、复旦大学浦东医学中心,上海究本科技有限公司
亮 点
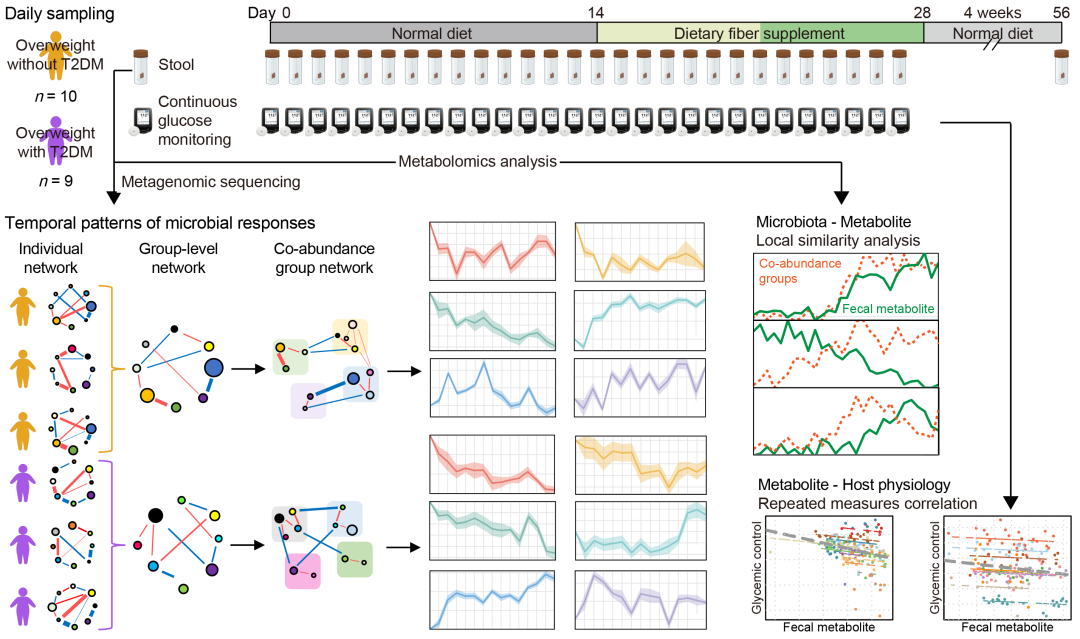
● 每日采样揭示了肠道菌群对膳食纤维的动态响应模式(这些模式在仅比较干预前后两个时间点时常被忽略),并将这些模式与微生物碳水化合物代谢的遗传能力相关联;
● 基于个体网络的功能群分析揭示了共现微生物成员间的稳定互作关系;
● 时滞效应分析发现了对宿主健康具有积极影响的微生物-代谢物关联,提示了潜在的因果关系验证目标。
摘 要
肠道菌群是一个高度动态且复杂的生态系统,其成员对膳食纤维的响应过程仍未被完全阐明。本研究对超重受试者(含2型糖尿病患者与非糖尿病患者)进行了连续28天的每日采样,包括前14天的习惯饮食观察期及后14天的膳食纤维干预期。结合微生物功能群划分与时间序列分析,我们揭示了不同菌群成员的多种时间响应模式,这些模式在传统采样方法中往往会被遗漏。我们还发现,时间响应模式与菌群成员碳水化合物利用和运输的遗传能力密切相关。此外,通过纵向宏基因组和代谢组数据的时滞效应分析,我们发现了可能介导肠道菌群对宿主代谢有益效应的特定代谢物。总体而言,我们的研究结果证明了高频采样对捕捉肠道微生物动态响应的必要性,并为机制研究提供了可靠靶点。
视频解读
Bilibili:https://www.bilibili.com/video/BV1nHEqzBEkd/
Youtube:https://youtu.be/ZKTV1SzDOHg
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
靶向肠道菌群的膳食纤维干预被广泛用于改善人类健康。这些有益效果是通过不同肠道菌群成员之间复杂的交叉喂养作用实现的。该过程涉及初级降解菌将复杂多糖水解为单糖或寡糖,随后由后续营养级的成员利用并发酵产生短链脂肪酸(SCFAs)。其中,识别能够利用膳食纤维的关键菌群成员及其随时间变化的动态响应是一项重要的研究内容。先前研究已取得一定进展:Liu等通过构建生态模型,确定了小鼠肠道菌群中菊粉的初级降解菌;类似地,Wei等通过¹³C标记,发现了小鼠肠道中菊粉诱导的Parabacteroides distasonis的富集;此外,Gerber等开发了用于分析纵向宏基因组数据的模型,成功识别出健康受试者在聚糖干预期间表现出相似动力学响应的微生物群。然而,不同研究结果之间的差异以及机制验证的缺乏,限制了我们对肠道菌群动态的理解。
当前大多数饮食干预临床研究采用横断面设计或仅设置有限的采样点。由于菌群对环境变化反应迅速(饮食调整后24小时内即可发生改变),这些传统采样方法无法完整捕捉试验期间菌群的变化。并且,肠道菌群的变化具有时间累积效应。这一点可从跨代积累的遗传适应性中得到证实,例如在Bacteroides thetaiotaomicron中,标准饮食和西方饮食诱导的适应性突变仅需两周即可积累。理论上,宿主生理变化反映的是整个干预期间微生物动态变化的累积结果,而非仅由干预结束时的菌群决定。这些基本生物学原理表明,研究肠道菌群的动态变化需要采用高频采样。此外,整合纵向多组学数据对于识别关键微生物至关重要,为后续揭示因果关系提供了基础。这种采样方法在环境微生物研究中已被证明是有效的,例如Seitz等通过每日采样阐明了覆盖作物根系的分泌物如何调控土壤微生物的氮循环和植物激素合成。
理解肠道菌群的动态变化还需认识其作为复杂适应系统的本质特征。在这个生态系统中,微生物成员相互作用并自发形成功能单元(也称为共丰度群,CAGs),共同影响宿主代谢。同一功能单元中的成员对环境变化(特别是营养变化)会表现出一致响应,即协同增长或衰减。值得注意的是,执行关键生物功能的菌群成员往往表现出稳定的相互作用,即在不同时间和生理条件下持续表现为正向或负向关联。此外,尽管微生物组数据固有的高维稀疏性对应用各种统计模型构成了挑战,基于功能群的分析提供了一种有效的解决方案。例如,Wu等人展示了如何有效地将约200万基因缩减至161个基因组,并进一步聚类为18个功能群,从而成功识别出菌群-疾病表型关联,并构建稳健的预测模型。因此,基于生态功能群的分析方法不仅符合肠道菌群的生态学特性,也提供了高效的降维策略,对探索微生物-宿主相互关系至关重要。
本研究选择膳食纤维作为干预因素,在19名超重受试者(包括2型糖尿病患者)中开展纵向临床试验。通过每日收集粪便样本监测肠道菌群组成和代谢物变化,并采用连续血糖监测仪追踪受试者血糖波动情况。本研究构建了包含每日采样、时间序列分析和共丰度群分析的综合分析框架,以更高的时间分辨率检测了菌群的动态响应。我们还进一步表征了各类菌群成员在碳水化合物利用/转运方面的功能基因特征。此外,基于纵向数据的时滞分析,我们发现了两组可能的“微生物-代谢物-宿主代谢”相互作用关系。总体而言,本研究深化了我们对肠道菌群动态变化的认识,为后续机制研究提供了可靠靶点。
结 果
膳食纤维快速引起血糖改善及微生物群变化
2021年8月至10月,我们招募了20名超重受试者,其中10人患有2型糖尿病(T2DM)。基线时两组受试者体重指数(BMI)相当(超重组:27.78 ± 1.12 kg/m²,T2D组:27.75 ± 0.80 kg/m²)。除空腹血糖(FPG)和糖化血红蛋白(HbA1c)水平外,其余指标在组间均无显著差异。本研究包括两周观察期(受试者保持常规饮食,第0-13天,图1A)、两周膳食纤维补充期(第14-27天)及四周随访期(第28-56天)。膳食纤维补充期分为低剂量阶段(第14-20天,18 g/天)和高剂量阶段(第21-27天,36 g/天)。T2DM患者在整个试验期间持续服用降糖药物。超重组全部10名受试者和T2D组9名受试者完成了试验。
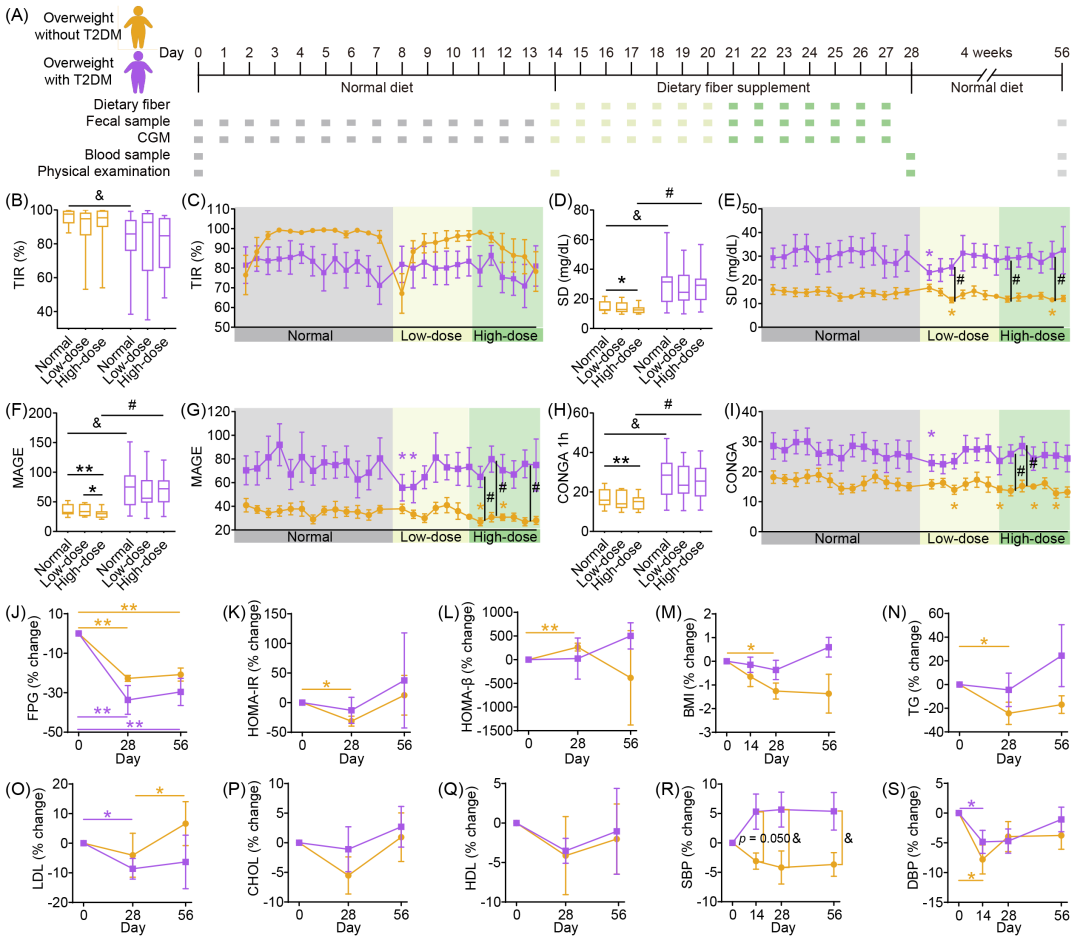
图1.膳食纤维快速改善受试者血糖稳定性
(A)研究设计与采样方案。研究包含2周常规饮食期、2周膳食纤维干预期和4周随访期。(B-I)试验期间血糖在70至180 mg/dL范围内的时间百分比(B和C)、日平均血糖标准差(D和E)、平均血糖波动幅度(F和G)、连续重叠净血糖作用(H和I)的变化。箱线图显示中位数和四分位距(IQR),须线延伸至最小值和最大值。折线图数据以均值 ± 标准误(SEM)展示。(J-S)受试者空腹血糖(J)、胰岛素抵抗指数(K)、胰岛素分泌指数(L)、体重指数(M)、甘油三酯(N)、低密度脂蛋白(O)、总胆固醇(P)、高密度脂蛋白(Q)、收缩压(R)和舒张压(S)的变化。
本研究将每日血糖处于70-180 mg/dL范围内的时间百分比(TIR)作为主要结局指标。膳食纤维干预期间,超重组和T2D组的TIR均维持在70%以上(符合糖尿病管理标准),但与常规饮食期相比无显著变化(图1B、C)。次要结局指标包括血糖变异性(GV)、血脂、血压和BMI。GV评估采用标准差(SD)、平均血糖波动幅度(MAGE)和连续重叠净血糖作用(CONGA)指标。与常规饮食期相比,膳食纤维干预期间两组GV均降低(图1D-I)。每日数据分析显示,膳食纤维补充开始后两组受试者GV均迅速显著下降(图1E、G、I)。膳食纤维补充还显著降低了两组受试者干预结束(第28天)和随访时(第56天)的空腹血糖(FPG,图1J)。超重组在胰岛素抵抗指数(HOMA-IR)、胰岛素分泌指数(HOMA-β)、BMI和甘油三酯方面改善更为显著(图1K-N),而T2D组的低密度脂蛋白(LDL)下降更明显(图1O)。其他次要结局指标无变化(图1P-S)。
通过对433份粪便样本进行鸟枪法宏基因组测序,并进行de novo组装,我们获得了1,100个高质量组装基因组(HQMAGs)。与常规饮食期相比,膳食纤维提高了两组受试者肠道菌群的多样性(Shannon指数)和丰富度(菌株数量,图2A、B),且这些变化在干预开始后三天内即表现出显著差异(图2C、D)。此外,膳食纤维还增强了α多样性的稳定性,表现为T2D组Shannon指数和超重组菌株数量变异系数的降低(图2E、F)。
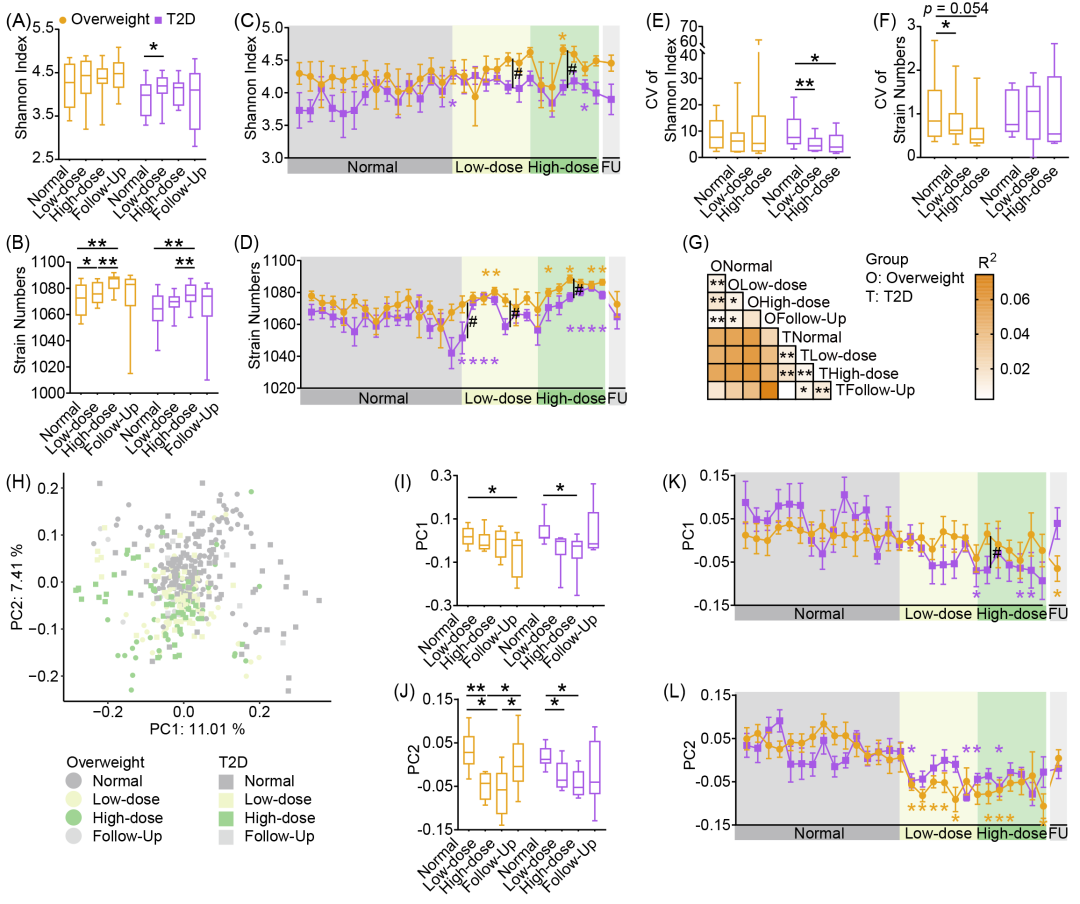
图2. 膳食纤维快速提高受试者肠道菌群α多样性并改变菌群结构
(A-F)肠道菌群多样性(Shannon指数)与丰富度(菌株数量)的变化。(A)各试验阶段Shannon指数。(B)各试验阶段菌株数量。(C)Shannon指数的每日变化。(D)菌株数量的每日变化。(E)Shannon指数的变异系数。(F)菌株数量的变异系数。(G)基于Bray-Curtis距离的分层PERMANOVA检验。(H)基于高质量组装基因组(HQMAG)水平Bray-Curtis距离的协变量校正主坐标分析(aPCoA),散点表示样本。(I-L)肠道菌群在PC1和PC2上的变化。(I)各试验阶段PC1。(J)各试验阶段PC2。(K)PC1的每日变化。(L)PC2的每日变化。
个体间差异解释了菌群组成变异的主要部分(75.03%),这与既往研究结果一致。在校正个体差异后,我们发现从常规饮食期到低/高剂量期,膳食纤维在两组中均诱导了菌群结构的显著变化(图2G-L)。每日采样数据显示,菌群变化最早出现在干预开始后的第二天(图2L)。总体而言,膳食纤维有效改善了受试者的糖代谢,同时快速提升了肠道菌群多样性并改变了其群落结构,这些变化始于干预初期。
肠道微生物对膳食纤维的动态响应模式
我们基于稳定的菌群成员间相互关系划分共丰度群(CAGs),从功能群层面探究了肠道菌群对膳食纤维的响应。基于个体共丰度网络,我们筛选了各组60%以上受试者中显著且一致的相关关系。总计在超重组选择了1,000个HQMAGs(占样本丰度的96.2 ± 0.25%),在T2D组选择了902个HQMAGs(占94.46 ± 0.30%),进一步分别划分得到82个CAGs和78个CAGs(图3A、B)。
与仅考虑干预前后菌群丰度变化不同,我们采用时间序列分析捕捉了肠道菌群在膳食纤维干预期间每日的变化。使用常规分析方法,我们观察到超重组11个CAGs和T2D组26个CAGs的丰度在干预后有显著变化。进一步,利用线性混合模型我们发现,超重组60个CAGs和T2D组69个CAGs在干预期间至少有一个时间点的丰度发生了显著变化(图3C、D)。这些发现凸显了纵向数据在捕捉更广泛、更全面的肠道菌群动态方面的优势。

图3. CAGs对膳食纤维展现多种时间响应模式
(A与B)超重组(A)和T2D组(B)试验期间的共丰度网络。仅保留组内至少六名受试者中持续显著(p ≤ 0.05)的正/负相关关系纳入共丰度网络。节点颜色表示HQMAGs的共丰度群(CAG)归属。为清晰展示,仅绘制超重组中相关系数绝对值 ≥ 0.7、T2D组中 ≥ 0.6的边。(C与D)热图显示膳食纤维干预期间CAG丰度变化,均以常规饮食期均值为基准。(E–H)热图展示超重组(E与G)和T2D组(F与H)中各Cluster的CAZy基因(E-F)及磷酸转移酶系统(PTS)KEGG直系同源基因(KOs,G-H)平均占比。
基于动态时间规整距离聚类,我们在超重组识别出8种时间响应模式,其中Cluster G和H在干预期间丰度保持稳定(图3C)。Cluster D、E和F的丰度在干预期间增加,主要由已知可利用碳水化合物产生短链脂肪酸(SCFAs)的物种组成。Cluster D的丰度从干预第二天起持续显著增加,其中3个CAGs的主导菌来自Bifidobacterium(B. longum和B. pseudocatenulatum)。Bifidobacterium可通过独特的“bifid shunt”途径高效转化碳水化合物为ATP,在富含膳食纤维的环境中快速增殖。此外,Cluster D中还包含6个由厚壁菌门中常见的SCFAs产生菌主导的CAGs,优势物种包括Agathobacter faecis、Blautia_A massiliensis、Fusicatenibacter saccharivorans、Faecalibacterium prausnitzii_G、F. prausnitzii和Roseburia hominis。Cluster E的丰度在低剂量阶段显著增加,随后在高剂量阶段下降。Cluster E的16个CAGs中,12个是由毛螺菌科(Lachnospiraceae)的SCFAs产生菌主导,包括Anaerostipes hadrus、Mediterraneibacter faecis、Roseburia inulinivorans、Agathobacter faecis、Anaerobutyricum hallii、Anaerobutyricum soehngenii和Bariatricus comes。该Cluster另外4个CAGs由可产乙酸/乳酸的Blautia_A wexlerae和Blautia_A faecis主导,这些菌可通过交叉喂养支持其他丁酸产生菌生长。Cluster F的丰度在整个干预期间呈上升趋势,其11个CAGs中6个由厚壁菌门的丁酸产生菌主导,包括Agathobacter rectalis、Agathobaculum butyriciproducens、F. prausnitzii_G、F. prausnitzii_C和Clostridium_A leptum。Cluster D、E和F主要由厚壁菌门的SCFAs产生菌主导,这一点与厚壁菌门成员具有更强的聚糖结合能力,在膳食纤维富集环境中具有竞争优势相符。此外,尽管存在这些相似性,各Cluster间不同的时间响应模式可能源于主导菌种在碳水化合物代谢酶和环境适应能力方面的物种特异性差异。
超重组Cluster A、B和C的丰度在干预期间降低。Cluster A在低剂量阶段显著下降,以pH敏感的Bacteroides和潜在病原菌或有害菌(Klebsiella pneumoniae、Parasutterella excrementihominis和Enterocloster bolteae)为主。此外,Cluster A其余的4个CAGs具有较高物种多样性。Cluster B的丰度从干预第二天起持续下降,以酸性环境敏感的Bacteroides spp.和Phocaeicola vulgatus为主导菌。Cluster C的丰度主要在高剂量阶段下降,其4个CAGs由多种与病理状态相关的物种主导,包括Bilophila wadsworthia、Clostridium_AQ innocuum、Escherichia coli_D和Haemophilus_D parainfluenzae。Cluster C另有两个CAGs显示较高物种多样性。这些发现表明,丰度下降的CAGs具有pH敏感性、潜在致病性或物种多样性较高等特征。
时间序列分析结果与仅比较干预前后两个时间点的分析结果存在差异。尽管后者识别出7个干预后丰度增加的CAGs,但遗漏了Cluster E在低剂量阶段的短暂增加,并将Cluster E内的CAG 5和9错误判断为干预后丰度显著下降。此外,这种分析方法仅发现2个丰度降低的CAGs,忽略了其余丰度降低的变化模式。这些结果凸显了每日采样和时间序列分析能够更全面地表征微生物动态变化的方法学优势。
在T2D组中,我们识别出8种时间响应模式,其中许多与超重组相似(图3D)。我们观察到Cluster H的丰度在干预期间稳定。在丰度增加的Cluster中,T2D组的Cluster E和超重组的Cluster D均呈现快速且持续的增长,其中包含多种SCFAs产生菌(Bifidobacterium pseudocatenulatum、F. prausnitzii、F. prausnitzii_G、Fusicatenibacter saccharivorans和Roseburia hominis)。类似地,T2D组的Cluster F和超重组的Cluster E的丰度均在低剂量阶段初期增加,均含有Anaerobutyricum soehngenii和Blautia_A wexlerae。而T2D组的Cluster G与超重组的Cluster F虽然均呈现丰度上升趋势,但其优势菌种组成不同。在丰度降低的Cluster中,T2D组的Cluster A与超重组的Cluster B均呈现快速下降趋势,两者都含有pH敏感的Phocaeicola vulgatus。此外,T2D组的Cluster D与超重组的Cluster A均呈现丰度先降后升的变化模式。总体而言,在两组受试者中观察到的相似时间响应模式和系统发育特征表明,肠道微生物对膳食纤维的响应在不同宿主之间表现出一致性。
功能基因分析进一步揭示,具有不同时间响应模式的菌群成员在碳水化合物代谢相关基因潜力方面存在差异。在碳水化合物利用方面,两组中丰度增加的Cluster均表现出更高比例的淀粉、菊粉和蔗糖/果聚糖利用相关基因(图3E、F)。在转运能力方面,这些Cluster也显示出更高比例的磷酸载体蛋白、磷酸烯醇式丙酮酸-蛋白磷酸转移酶和β-葡萄糖苷转运蛋白(图3G、H)基因。相反,丰度降低的Cluster具有更高比例的果胶利用和半乳糖醇/氮转运相关基因。上述结果表明,丰度变化趋势相近的菌群成员在碳水化合物代谢和转运方面呈现一致性,这与其分类学相似性相符。
肠道菌群与粪便代谢物在血糖调节中的时序关系
全局非靶向代谢组学分析显示,粪便代谢谱的个体间变异性低于肠道菌群结构的差异,提示尽管菌群组成存在个体差异,菌群功能在受试者间仍具有一定程度的相似性。与常规饮食阶段相比,两组的粪便代谢物谱在干预阶段均表现出显著变化(图4A-D),且这种变化最早在低剂量阶段首日即可观察到(图4E,F)。基于受试者干预期间浓度增加的代谢物进行通路富集分析,我们发现低剂量膳食纤维干预显著上调了多数受试者的氨基酸代谢(图4G,H)。值得注意的是,与低剂量干预相比,高剂量膳食纤维干预进一步上调了3名非糖尿病超重受试者的维生素B6代谢(图4I,J)。高剂量干预还显著上调了两组中的多条代谢通路,尤其是苯丙氨酸和色氨酸代谢相关通路(图4K,L)。综上所述,膳食纤维能够快速且显著地诱导受试者粪便代谢谱发生变化。
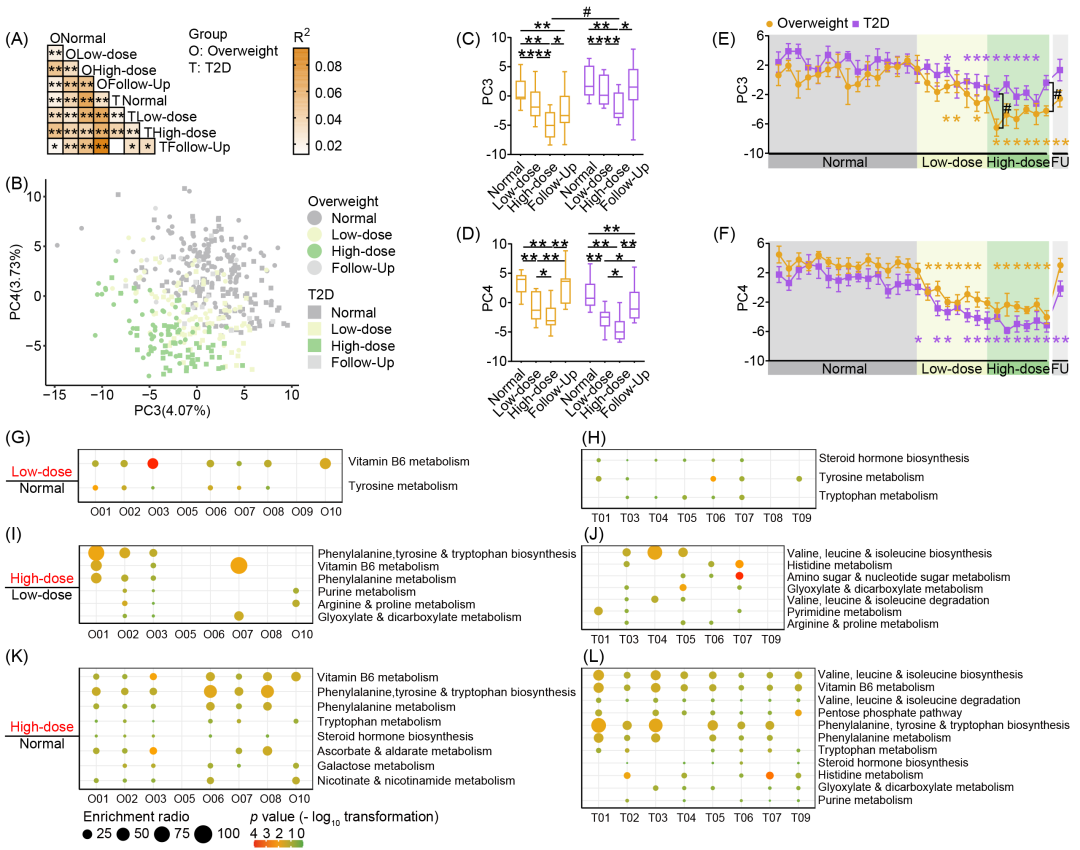
图4. 膳食纤维快速改变受试者粪便代谢组特征
(A)PERMANOVA(基于欧氏距离,999次置换,BH校正p < 0.05)。(B)粪便代谢物主成分分析(欧氏距离),散点代表样本。(C-F)PC3与PC4变化。(C)各试验阶段PC3。(D)各试验阶段PC4。(E)PC3的每日变化。(F)PC4的每日变化。(G-L)基于不同饮食阶段富集的粪便差异代谢物的通路富集结果:超重组(G,I,K)与T2D组(H,J,L)。(G,H)低剂量干预期与常规饮食期相比富集的通路。(I,J)高剂量干预期与低剂量干预期相比富集的通路。(K,L)高剂量干预期与常规饮食期相比富集的通路。
Procrustes分析证实了超重组和T2D组粪便代谢物谱与菌群组成之间存在显著相关性。在超重组中,25个显著的时滞相关关系提示CAGs可能参与了相应物质的产生或代谢调控(图5A)。值得注意的是,CAG80的丰度变化与4-吡哆酸(4-PA,维生素B6的主要代谢产物)的浓度变化呈现显著正相关(局部相似性评分 = 0.760;延迟 = 3天;FDR = 0.044),且二者均在干预开始后持续增加(图5B,C)。CAG80中全部的7个HQMAGs均包含编码4-PA代谢关键酶的基因(图5D,E)。此外,4-PA与受试者平均血糖(MBG)、血糖管理指标(GMI)、夜间血糖标准差(Nighttime SD)以及J指数呈显著负相关(图5F)。
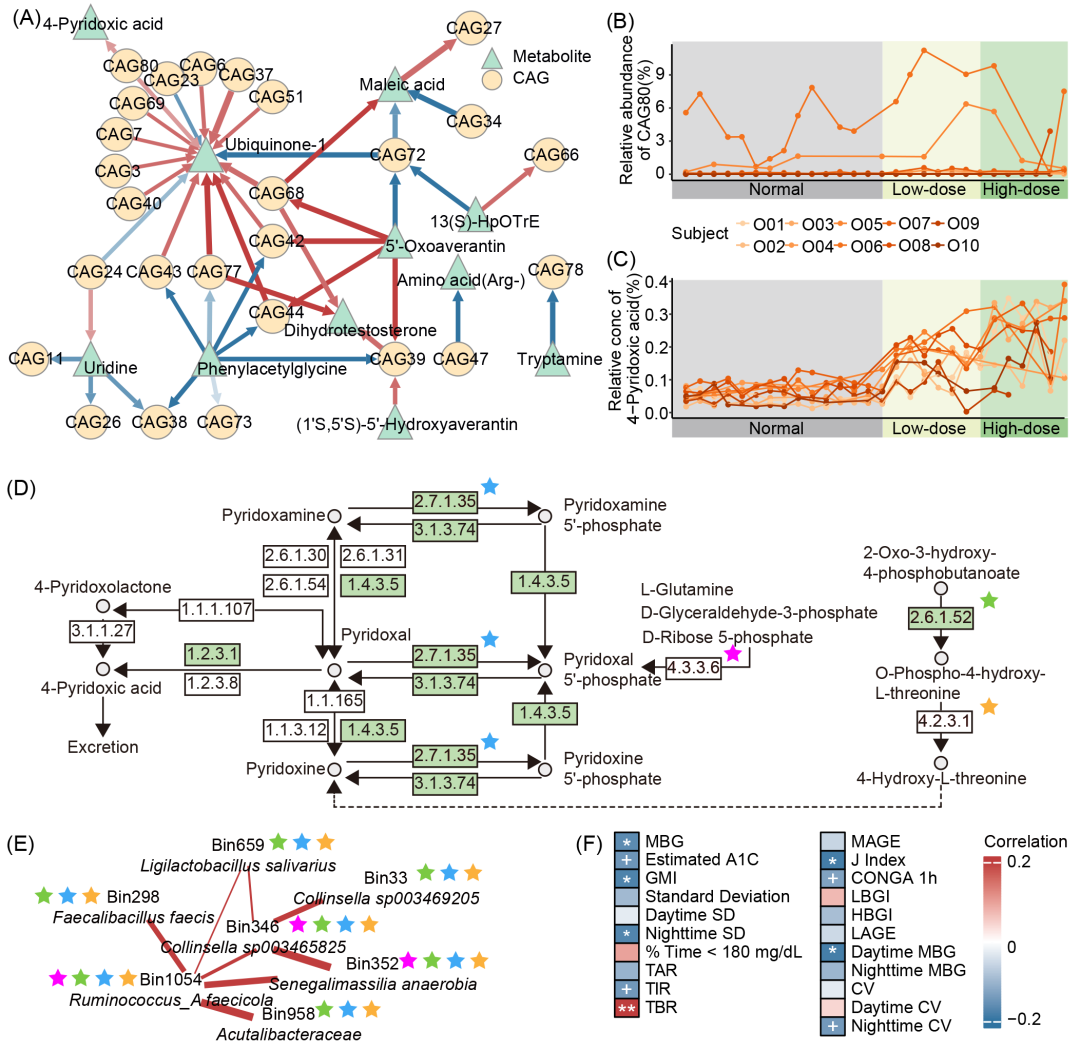
图5. 4-吡哆酸在超重组肠道菌群与宿主血糖稳定性关联中的作用
(A)超重组CAGs与粪便代谢物的显著时滞相关性。箭头表示时滞关系方向,红色表示正相关,蓝色表示负相关。线宽代表局部相似性评分绝对值,颜色深浅反映节点间时滞时长,颜色越深表示时滞越短。(B与C)超重组CAG80丰度变化(B)与4-吡哆酸水平变化(C)。(D)维生素B6代谢通路(KEGG #00750)。可在超重组CAG80的HQMAGs基因组中注释到的KOs以不同颜色星号标识,人类基因组编码的KOs以绿色表示。(E)超重组CAG80所包含的HQMAGs间相互作用关系。边宽度表示相关系数绝对值,红色表示正相关。星号代表HQMAGs基因组中注释到的KOs。(F)4-吡哆酸与连续动态血糖监测(CGM)指标的重复测量相关性。红色表示正相关,蓝色表示负相关。
类似地,在T2D组中也发现25个显著的时滞相关关系表明CAGs对代谢物具有潜在调控作用(图6A)。其中,CAG8与马来酸呈正相关(局部相似性评分 = 0.729;延迟 = 1天;FDR = 0.046),且两者在干预期间均快速增加(图6B,C)。CAG8中来自Blautia obeum和Blautia faecis的两个HQMAGs基因组均包含编码马来酸代谢关键酶的基因(图6D,E)。此外,马来酸水平与受试者的日间平均血糖呈负相关(图6F)。这些结果表明,响应膳食纤维干预的肠道菌群成员可能通过调控特定代谢物,进而改善宿主的血糖控制。
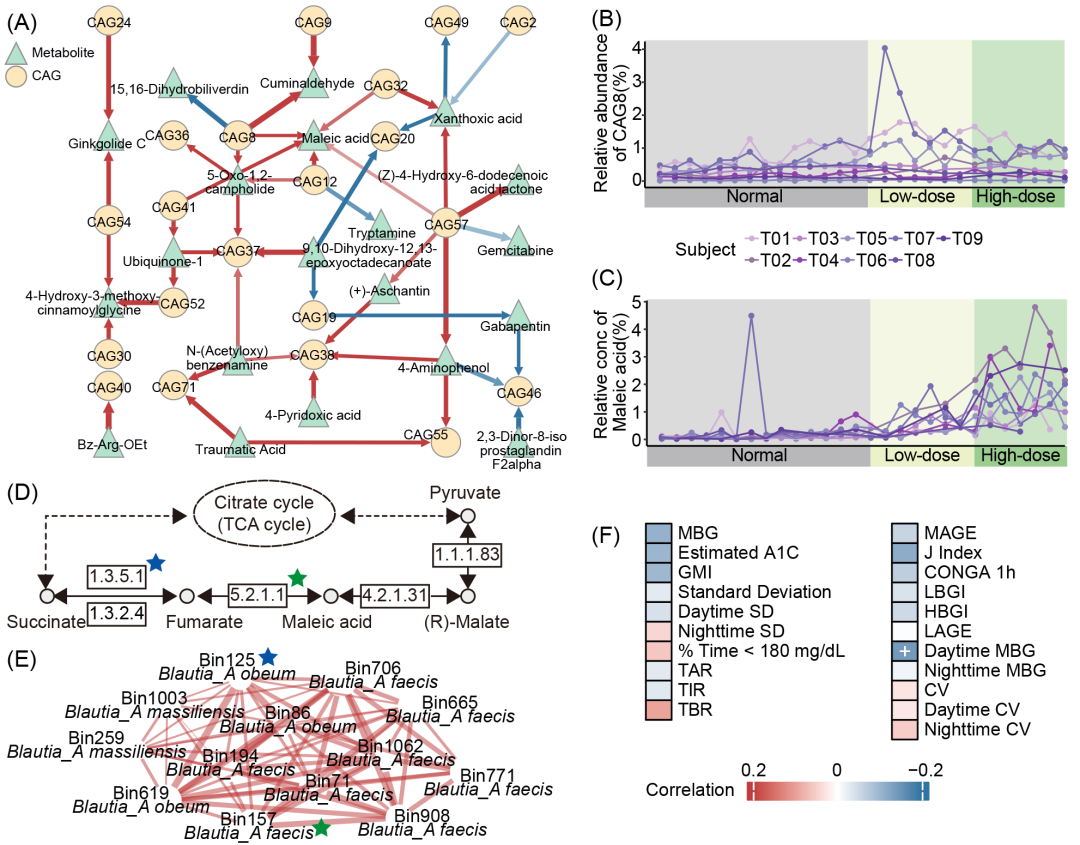
图6. 马来酸在T2D组肠道菌群与宿主血糖稳定性关联中的作用
(A)T2D组CAGs与粪便代谢物的显著时滞相关性。(B与C)T2D组CAG8丰度变化(B)与马来酸水平变化(C)。(D)丁酸代谢通路(KEGG #00650)。T2D组CAG8的HQMAGs基因组中注释到的KOs以不同颜色星号标识。(E)T2D组CAG8所包含的HQMAGs间相互作用关系。星号代表HQMAGs基因组中注释到的KOs。(F)马来酸与CGM指标的重复测量相关性。
血清代谢物在肠道菌群改善宿主代谢过程中的介导作用
与基线(第0天)相比,在膳食纤维干预结束时(第28天)和随访期(第56天),两组受试者的血清代谢物谱均发生了一定程度的变化(图7A,B),并且在超重组和T2D组中分别检测到23和34个差异代谢物(图7C,D)。代谢通路富集分析显示,牛磺酸/次牛磺酸代谢通路和初级胆汁酸生物合成通路在两组中均呈现显著上调(图7E)。此外,多条代谢通路在粪便和血清代谢组中均呈现共同上调趋势,其中超重组中包括类固醇激素生物合成和色氨酸代谢通路,T2D组中则包括维生素B6代谢及乙醛酸/二羧酸代谢通路。
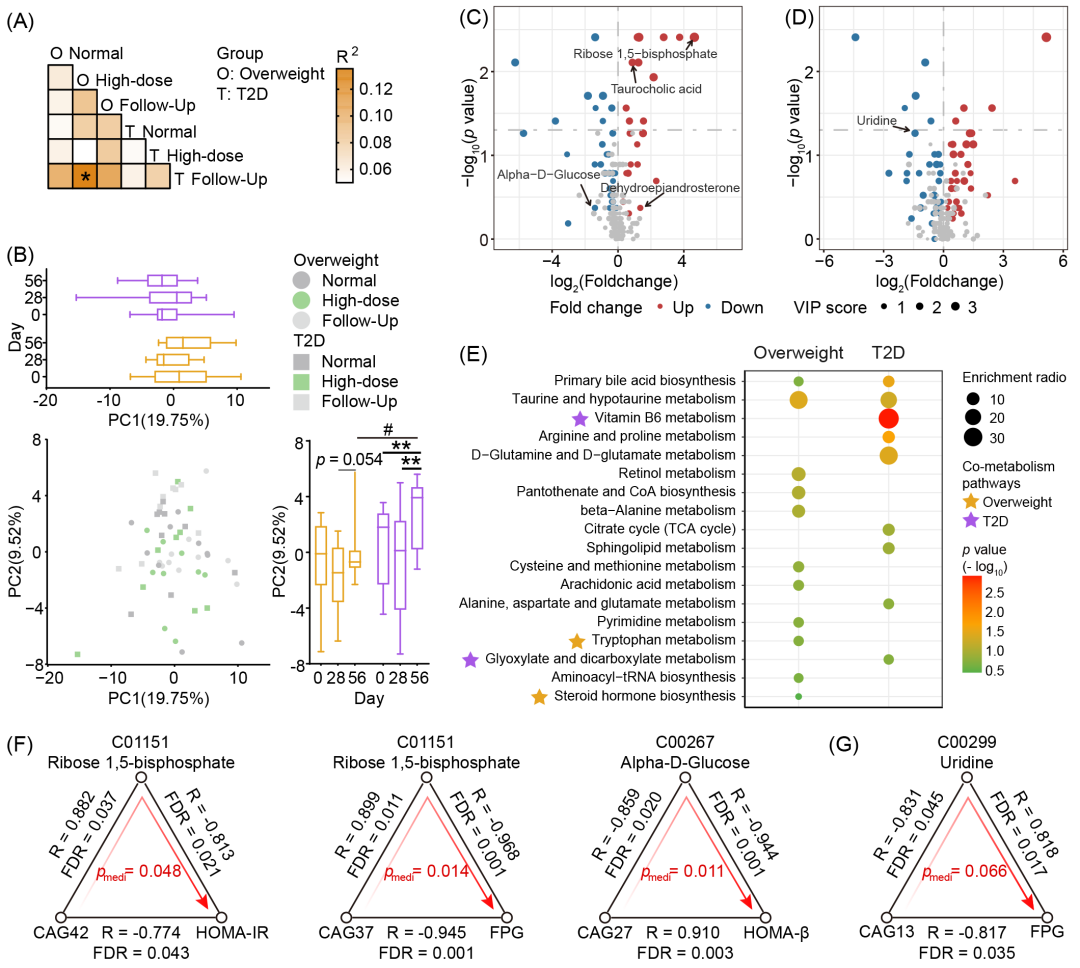
图7. 膳食纤维干预后血清代谢组的显著变化
(A)基于欧氏距离的PERMANOVA(999次置换,BH校正p < 0.05)。(B)血清代谢物水平主成分分析(欧氏距离),散点代表样本。PC1与PC2的变化通过箱线图展示。(C与D)火山图显示超重组(C)和T2D组(D)干预前后血清差异代谢物,变量重要性投影(VIP) ≥ 1的代谢物以红/蓝色标识。(E)基于两组中膳食纤维干预后上调的血清差异代谢物的通路富集分析。星号标注在粪便与血清代谢组中共同富集的代谢通路。(F与G)超重组(F,p < 0.05)和T2D组(G,p < 0.1)中血清代谢物的中介效应。
中介效应分析结果显示超重组中存在6个显著关联(图7F)。其中,Ribose 1,5-bisphosphate(Rib-1,5-P2)可以介导CAG42对胰岛素抵抗指数(HOMA-IR)及CAG37对空腹血糖的影响。功能基因分析显示,CAG42(以假小链双歧杆菌Bifidobacterium pseudocatenulatum为主)和CAG37(以食糖梭杆菌Fusicatenibacter saccharivorans为主)均具备通过磷酸戊糖途径合成Rib-1-P(Rib-1,5-P2的上游代谢物)的能力。同时,Alpha-D-Glucose可介导CAG27对胰岛素分泌功能指数(HOMA-β)的影响。以普拉梭菌(Faecalibacterium)为主的CAG27具备通过多种葡萄糖代谢途径影响Alpha-D-Glucose代谢的能力。在T2D组中,尿苷可能介导CAG13对空腹血糖的调节作用(p < 0.1,图7G)。功能基因注释结果表明,以Blautia_A为主的CAG13具备通过ABC转运系统和嘧啶代谢通路调控尿苷代谢的潜能。这些发现为理解“肠道菌群-血清代谢物-宿主代谢”的相互作用提供了新的见解。
讨 论
我们的研究强调了高频采样在肠道菌群动态研究中的必要性,而这一点常被传统采样方法所忽视。以膳食纤维作为有效扰动因素,我们整合了每日采样、功能群分析和时间序列分析,揭示了具有不同碳水化合物代谢遗传能力的微生物成员的独特响应模式。对纵向组学数据的时间延迟分析,揭示了可能介导响应膳食纤维的肠道菌群成员与宿主代谢改善相互作用的具体代谢物。总体而言,我们建立的综合分析框架为机制研究提供了可靠靶点,并为精准治疗干预奠定了基础。
高频采样在本研究中展现出准确表征微生物动态的显著优势。关于微生物动态的识别,我们的对比分析表明,仅依赖干预前后的采样数据可能导致误导性结论。例如,尽管干预前后两个时间点的比较显示超重组中CAG5和CAG9丰度降低,但每日采样数据表明,这些CAGs在低剂量干预阶段早期实际显著增加。此类误判可能导致部分肠道菌群成员的潜在生物学意义被忽视。具体而言,CAG5中的Mediterraneibacter faecis已知在多糖降解和SCFAs合成中发挥关键作用。类似地,CAG9中的优势物种Anaerobutyricum soehngenii能够促进超重和胰岛素抵抗受试者胰岛素敏感性的提高。这些发现表明,传统采样方法可能会遗漏具有重要功能意义的瞬时微生物动态变化。
高频采样能够揭示组学数据之间的时间延迟相关性。通过局部相似性分析,我们发现了肠道菌群、代谢物与宿主代谢之间的潜在相互作用。关于在超重组中发现的4-PA,先前在ob/ob小鼠中的研究已证明其具有改善葡萄糖代谢的能力。类似地,在T2D组中发现的马来酸的潜在代谢益处与Zucker糖尿病肥胖大鼠的研究结果一致。在这些大鼠中,盲肠马来酸浓度与血浆丙氨酸转氨酶(ALT)水平呈负相关。综上所述,这些研究发现表明,高频采样可为机制研究和新型治疗策略的开发提供更为准确和可靠的靶点。
菌株水平分析对准确表征微生物对饮食干预的响应至关重要。与同类研究的比较显示,尽管来自Bacteroides属的菌种具有快速响应饮食干预的共同特征,但其在人类与动物模型中的时间动态模式存在差异。在本项研究中,Bacteroides uniformis是超重组中CAG25以及T2D组中CAG6和CAG7的优势物种,这些CAGs的丰度在膳食纤维干预期间降低。相比之下,使用动物模型进行的研究发现,菊粉干预后B. uniformis的丰度显著增加。这种差异可能反映了同一物种在不同宿主中的基因组变异,提示微生物对干预的响应可能具有菌株特异性。因此,未来研究应聚焦于更为详细的菌株水平分析,而非仅限于属或种水平的研究,以更全面地捕捉微生物对饮食干预的动态变化。
基于功能群的聚类分析是理解肠道微生物成员间相互作用的稳健方法。通过聚焦于稳定的成员间相互作用,该方法部分克服了传统基于分类单元和基于基因的方法的局限性。该方法的有效性和广泛适用性已在多项研究中得到验证,这些研究中识别出的功能协同的微生物群,与肥胖、2型糖尿病和COVID-19等多种疾病显著相关。并且,研究人员在不同疾病中均观察到了功能群之间竞争性的“跷跷板”结构,这进一步凸显了该方法在疾病分类和干预效果评估中的应用价值。值得注意的是,本研究通过捕捉菌群成员间的真实物理共存关系,进一步提高了CAG聚类分析的可靠性。我们通过构建个体微生物网络并仅保留每组至少6名受试者中一致识别到的相关性,有效减少了由个体间差异引起的虚假关联。功能分析进一步证实了我们聚类结果的生物学意义:具有不同时间动态模式的CAGs呈现出差异化的碳水化合物利用和物质转运相关基因特征。此外,CAG动态变化、代谢物水平变化与宿主血糖变异性之间的显著相关性,为我们的聚类方法提供了验证。总而言之,这些发现共同证明了,将微生物间物理共存关系纳入分析可提高结果的可靠性。
基于上述方法,我们揭示了微生物成员间相似时间响应模式的遗传基础。功能基因的相似性表明这些微生物具有相近的代谢功能。就碳水化合物代谢而言,丰度增加的CAGs中,与菊粉和蔗糖/果聚糖利用相关的CAZy基因占比更高,这些基因主要来自四个糖苷水解酶(GH)家族。具体而言,GH32家族包含能够水解果聚糖糖苷键的糖苷水解酶;GH91家族的菊粉果糖基转移酶可将菊粉转化为双果糖酐III;GH70家族可利用蔗糖和淀粉合成α-葡聚糖;GH68家族优先以蔗糖为底物合成果聚糖和果寡糖。在物质转运方面,丰度增加的CAGs中β-葡萄糖苷转运相关基因占比更高。由bglF和bglP编码的PTS系统EIIBCA组分负责胞外β-葡萄糖苷的转运和磷酸化。先前研究表明,这些转运组分的缺失会显著影响β-葡萄糖苷、蔗糖和果糖等多种糖的利用,凸显了它们在物质转运中的关键作用。然而,需要注意的是,基因比例的分布不一定能够准确反映细菌内实际的酶活性。因此,多组学分析和实验验证对于确定微生物成员的精确代谢特性是必不可少的。
我们的研究也存在一些局限性。尽管本研究的队列规模相较于许多临床试验较小,但我们采用的每日采样策略和自身对照设计揭示了传统方法所忽视的肠道菌群动态变化模式。由于缺乏可比较的数据集,我们研究结果的外部验证面临挑战。此外,仅凭时间相关性无法确立因果关系,还需要通过实验进一步验证。作为未来发展方向,我们计划在多个研究中心开展大规模人群高频采样研究,以提高结果的可重复性和普适性。
结 论
本研究证实了高频采样在捕捉肠道菌群动态响应方面具有重要意义,而这些响应常常会被仅考虑干预前后两个时间点的比较所忽视。通过识别肠道菌群与有益宿主健康的代谢物之间的关联,该方法为因果关系研究提供了潜在靶点,并提高了肠道菌群成员与代谢物在临床中的应用潜力。未来研究可进一步探索该方法在多种疾病模型中的应用价值,从而深化对肠道菌群动态变化机制的理解。
方 法
临床研究注册和设计
本临床试验于2021年8月至10月在上海浦东医院和上海市第一人民医院开展,旨在探究超重受试者(包括2型糖尿病患者)在膳食纤维干预期间肠道菌群变化特征。研究方案经中国临床试验注册伦理委员会批准(ChiECRCT20210194,2021年6月20日)。研究遵循赫尔辛基宣言原则,并注册于中国临床试验注册中心(ChiCTR2100046478)。所有受试者均签署知情同意书。
共筛查23名肥胖/超重受试者(BMI 24.0–35.0 kg/m²,近三月体重变化 < 15%),年龄25–60岁,血压 < 180/110 mmHg,甘油三酯 < 8 mmol/L。根据近6个月体检报告或医疗记录符合T2DM诊断标准的受试者纳入T2D组。糖尿病诊断标准包括:HbA1c ≥ 6.5%(检测方法经美国国家糖化血红蛋白标准化计划认证,并严格遵循美国糖尿病控制与并发症试验流程);或任意时间点随机血糖 ≥ 11.0 mmol/L。
入组筛选包含病史采集、食物过敏评估、体格检查、OGTT及多项血液检测。排除标准包括:(1)妊娠、哺乳或计划怀孕;(2)糖尿病病程 > 2年;或 < 2年但伴并发症或低血糖;(3)严重感染、重大创伤、手术或其他急性应激状态;近1月内存在慢性胃肠道问题;(4)近3个月内使用噻唑烷二酮类、葡萄糖苷酶抑制剂、减肥药、皮质类固醇、影响胃肠动力的药物或移植后用药;(5)慢性肾病或血尿素氮或肌酐异常;慢性肝病或ALT或AST升高;血或尿淀粉酶升高;(6)严重或不稳定型心绞痛、冠状动脉供血不足或NYHA III/IV级心衰;糖尿病性周围神经病变伴疼痛、尿或胃潴留、足溃疡或需紧急处理的视网膜病变;(7)近1年内存在药物或酒精成瘾;(8)研究者判定影响研究参与或评估的其他状况。
本研究采用非随机对照的自身对照研究设计,研究目的并非评估膳食纤维干预前后主要结局指标和次要结局指标的显著变化,而是探究干预期间受试者肠道菌群组成的动态变化,因此未进行样本量计算。原计划每组招募8–20名超重受试者(T2D与非T2D均衡分配)。经筛查的23名受试者中,20名入组(超重组n = 10,T2D组n = 10)。其中1名T2D组受试者提前退出,1名超重组受试者曾报告轻度腹泻但继续服用膳食纤维补充剂。
受试者接受为期2周的膳食纤维干预:在低剂量阶段(Day 14–Day 20)每日餐前摄入18克,在高剂量阶段(Day 21–Day 27)每日摄入36克,同时保持原有日常饮食。低剂量阶段的具体方案为:早餐前摄入6克不溶性纤维,午餐和晚餐前各摄入混合纤维6克(含3克不溶性纤维与3克可溶性纤维)。高剂量阶段将纤维摄入量加倍。干预结束后进行为期4周的随访。T2D组受试者在研究全程持续使用医生开具的抗糖尿病治疗方案。主要结局指标为每日血糖达标时间的百分比(TIR,70–180 mg/dL);次要结局指标包括动态血糖波动、血脂(胆固醇、甘油三酯、低密度脂蛋白与高密度脂蛋白)、血压及BMI。
膳食纤维补充剂
受试者从第14天开始服用由上海究本科技有限公司提供的膳食纤维补充剂,该补充剂经通标标准技术服务有限公司检测确认营养成分及微生物/致病菌污染安全性。不溶性膳食纤维补充剂包含:小麦麸皮、燕麦麸、薏苡仁、荞麦、青稞、奇亚籽、藜麦、黑麦、栗米和罗汉果提取物。可溶性膳食纤维补充剂包含:抗性糊精、菊粉、低聚果糖和低聚木糖。受试者被建议用水冲服这些补充剂。
样本和数据采集
血液样本采集于第0、28、56天在上海浦东医院进行。样本室温静置30分钟后,在4 ± 2℃下以2,500 g离心15分钟分离血清,用于临床检测和实验室存储。静脉血样在上海市第一人民医院进行血糖、血常规和血液生化检测。
在常规饮食和膳食纤维干预阶段,受试者每日使用提供的容器自行采集粪便样本。样本立即通过装有冰袋的保温箱运输至实验室进行质量评估,并存储于-80℃冰箱中。第56天采集的晨起样本立即置于干冰中,运送至医院后转移至实验室-80℃冷冻。
体格检查于第0、14、28、56天在上海浦东医院进行。身高和体重使用带有测高仪的数字体重秤测量。腰围测量于肋骨下缘与髂嵴间最窄处,臀围测量于臀部最宽处。血压采用欧姆龙HEM-7200电子血压计测量,测量前受试者静坐休息15分钟。
试验第0天在受试者左上臂安装CGM传感器,每15分钟记录一次血糖数据,持续14天。研究人员在第14天为受试者更换传感器,第28天移除传感器。血糖达标时间百分比(TIR)、平均血糖波动幅度(MAGE)和连续重叠净血糖作用(CONGA)等指标通过R包cgmanalysis计算。由于传感器安装、更换和移除可能影响数据准确性,且第28天空腹可能干扰CGM测量,所以相关操作当天的数据被排除。
糖化血红蛋白(HbA1c)通过G8全自动糖化血红蛋白分析仪检测。血浆葡萄糖、总胆固醇、总胆红素、高密度脂蛋白、低密度脂蛋白、AST、ALT、微量白蛋白和尿肌酐通过ADVIA Chemistry XPT系统测定。血清胰岛素通过ADVIA Centaur XP系统测定。
临床数据统计分析
使用R(4.1.2版本)进行统计分析。CGM指标:常规饮食阶段组间比较使用Mann-Whitney检验(双尾);膳食纤维干预期间组间比较使用单因素协方差分析(ANCOVA),以常规饮食阶段均值为协变量。超重组多数分析中n = 9,高血糖指数分析中n = 8;T2D组常规阶段分析中n = 9,低/高剂量阶段分析中n = 8。体格指标百分比变化:同时间点组间比较使用Mann-Whitney检验(双尾);不同时间点组内比较使用Wilcoxon配对符号秩检验(CGM指标单尾,体格指标双尾)。显著性阈值设为 p < 0.05。超重组多数比较中n = 10,第14天收缩压和舒张压分析中n = 9,第28天收缩压和舒张压分析中n = 8;第28天腰围、臀围和腰臀比分析中n = 9。T2D组所有分析中n = 9。
宏基因组测序
共收集443份粪便样本。使用QIAamp PowerFecal Pro DNA试剂盒提取,并通过Qubit 3.0荧光计配合Qubit dsDNA HS检测试剂盒定量。DNA完整性经1%琼脂糖凝胶电泳评估。文库构建:300ng DNA经KAPA Frag酶和KAPA Frag缓冲液酶切片段化,65℃末端修复及加A尾30分钟。加入接头储存液、连接缓冲液和DNA连接酶后,20℃孵育15分钟。文库经VAHTS DNA Clean Beads纯化与片段筛选。扩增后文库通过安捷伦2100生物分析仪配合高灵敏度DNA试剂盒进行片段分析,并通过StepOnePlus实时定量PCR系统定量。宏基因组测序采用Illumina Novaseq 6000平台进行双端150bp测序。
数据质量控制
使用Trimmomatic v0.39对原始数据进行处理:移除含超过35bp'N'碱基及接头序列超过10bp的低质量读长。clean reads通过Bowtie2 v2.4.1比对至人类参考基因组GRCh38。
从头组装与分箱
高质量序列经MEGAHIT v1.2.9进行de novo拼接,生成 ≥ 500bp的contigs。Binning流程:使用Bowtie2 v2.4.1将高质量读长比对至contigs,经Samtools v0.1.19转换为BAM文件。通过MetaBAT2 v2.15.2计算contigs序列的比对深度。单样本contigs使用MetaBAT2 v2.15.2进行binning。为增加bins的数量和完整度,合并所有样本contigs后再次使用MetaBAT2进行binning。宏基因组组装基因组(MAGs)经CheckM v1.1.2质控。Mash v2.2.2构建MAGs层级聚类树。经dRep v3.2.0去冗余后共筛选得到1100个高质量MAGs(HQMAGs)用于菌株水平分析。
丰度计算与分类地位鉴定
采用GTDB-Tk v1.3和GTDB R95参考数据库(2020年7月版)对HQMAGs进行物种注释。通过Bowtie2 v2.4.1比对clean reads至HQMAGs获取reads丰度,相对丰度定义为根据每个HQMAG基因组大小标准化后的、与HQMAGs基因组比对上的reads数。
肠道菌群功能分析
使用PRODIGAL v2.6.3预测HQMAGs基因。预测的编码基因经DIAMOND v0.9.9.110比对至KEGG(2019.10)和CAZys(2020)数据库。筛选最高得分比对结果进行后续分析。
肠道菌群网络构建与分析
选取各受试者样本中占比 > 45%的HQMAGs构建共丰度网络。基于HQMAG丰度,通过FastSpar计算基因组相关性。仅保留p ≤ 0.05、组内 ≥ 6名受试者共有的显著正/负相关关系纳入组水平共丰度网络。组水平网络使用Cytoscape v3.10.1可视化,个体网络使用Gephi v0.10.1展示。每对HQMAGs的相关性系数为该组内受试者共丰度网络中对应相关性系数的平均值。相关系数矩阵转换为距离矩阵(1-相关系数),经Ward.D2法层级聚类。PERMANOVA鉴定差异CAGs。
微生物组数据统计分析
α/β多样性统计检验方法同CGM指标。通过累加CAG内HQMAGs的相关编码基因数,比较不同时间模式Cluster(增加/减少/不变)的碳水化合物利用/转运能力。CAZy基因与KOs比例的三组间两两比较采用Mann-Whitney检验(双尾);组间差异使用Kruskal-Wallis检验与Dunn事后检验评估。基于Bray-Curtis距离的PERMANOVA分析HQMAG水平菌群差异。LinDA v0.2.0用于识别丰度在干预前后有显著变化的CAGs,受试者作为随机效应。膳食纤维干预阶段CAG丰度时序变化分析采用R包lme4 v1.1-35.1的lmer函数构建模型:lmer(log(CAG)~ day + (1 | participant))。干预期间每日CAG丰度变化量为固定效应系数与截距(常规饮食平均丰度)之和。动态时间规整(DTW)距离经R包dtw v1.23-1计算,基于丰度时序变化相似性与轮廓系数确定最佳聚类数。
粪便与血清样本前处理(代谢组学分析)
粪便样本(100 mg)与100 mg玻璃珠、600微升预冷甲醇(-20℃,含同位素内标)混合研磨超声。混合物离心(12,000 rpm,4℃,10分钟)后经0.22 μm滤膜过滤。取等量上清混合作为质控样。血清样本(100 µL)4℃解冻,加入400 µL预冷甲醇(-20℃)和100 µL同位素内标,同条件离心。上清浓缩干燥后,用150 µL 80%甲醇(-20℃)复溶,离心过滤。取10-25 µL各血清样本混合作为质控样。
代谢物检测
液相色谱分析采用Vanquish UHPLC系统。色谱分离使用Acquity UPLC HSS T3色谱柱(2.1×150 mm,1.8 μm),40℃柱温,0.25 mL/min流速。代谢物通过Orbitrap Exploris 120质谱仪电喷雾离子源检测。采用MS1与MS/MS(全扫描-ddMS2模式,数据依赖性采集)同步采集,动态排除重复离子。数据经Proteowizard和XCMS处理,导出化合物信息(m/z、保留时间、峰面积)。通过mzCloud、LipidMaps、人类代谢组数据库及自建PANOMIX数据库进行化学注释。
代谢物统计分析
共鉴定得到1194种粪便代谢物,经MetaboAnalyst 6.0用四分位距法过滤。数据标准化包括总和归一化、log10转换及Pareto缩放。代谢组差异通过PERMANOVA评估。差异代谢物统计与通路富集分析使用MetaboAnalyst(v4.0)。通过OPLS-DA,Mann-Whitney U检验(双尾)筛选差异代谢物(p < 0.05且VIP ≥ 1)。血清差异代谢物筛选标准为 VIP ≥ 1。
Procrustes分析
通过vegan v2.6.4的procrustes函数,对HQMAGs的aPCoA与代谢组PCA进行Procrustes分析。
局部相似性分析
采用前述线性混合模型评估CAG丰度/粪便代谢物浓度在28天研究期间的整体变化趋势。通过ELSA v1.0.2的lsa_compute函数计算CAG丰度与粪便代谢物的时滞相关性。p值经理论近似法估算,仅保留BH校正后p < 0 .05的显著相关性。时滞相关性网络使用Gephi v0.10.1可视化。CGM指标与粪便代谢物的重复测量相关性通过R包rmcorr v0.6.0计算。
中介效应分析
通过重复测量相关分析筛选CAGs-血清代谢物-血糖指标显著相关对(BH校正p < 0.05)。因样本内相关性,采用以受试者为随机截距的LMM替代线性回归。因果中介分析通过R包mediation v4.5.0的mediate函数,推断血清代谢物在CAGs与血糖表型关联中的中介效应。
代码和数据可用性:
宏基因组测序数据已存储于PRJCA026382项目下并公开可获取(https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA026382)。代谢组学数据已存储于OMIX数据库OMIX009789项目(https://ngdc.cncb.ac.cn/omix/release/OMIX009789)。method部分展示了本研究中应用的分析工具参数。使用的数据与脚本可在GitHub获取(https://github.com/linlin0026/imeta_1208/)。补充材料(图表、图形摘要、幻灯片、视频、中文翻译版本及更新材料)可通过在线DOI或iMeta Science官网查阅:http://www.imeta.science/。
引文格式:
Xiaotong Lin, Chaoxun Wang, Biao Liu, Yin Zhu, Rui Zhai, Chenhong Zhang. 2025. “Temporal response patterns of human gut microbiota to dietary fiber.” iMeta 4: e70046. https://doi.org/10.1002/imt2.70046
作者简介

林晓彤(第一作者)
● 上海交通大学生命科学技术学院硕士研究生。
● 研究方向为代谢受损个体肠道菌群对膳食纤维的动态响应过程。

王朝迅(第一作者)
● 上海市浦东医院(复旦大学附属浦东医院)内分泌科副主任医师。上海市医学会内分泌分会性腺学组成员,担任上海市浦东新区医学会骨质疏松专委会委员,上海市浦东新区医学会核医学专委会委员,中国生物物理学会会员,中国中医药研究促进会第一届糖尿病专业委员会委员,河北医科大学兼职副教授。
● 研究方向为糖尿病、骨质疏松、肥胖及其并发症发病机制的研究。曾以第一作者或通讯作者发现SCI和中文核心期刊论文,作为主编和编委参编著作,并获发明和实用新型专利。

张晨虹(通讯作者)
● 上海交通大学生命科学技术学院、微生物代谢全国重点实验室研究员,博士生导师。
● 入选第四批国家“万人计划”青年拔尖人才、国家自然科学基金“优青”、仲英青年学者、上海市教委曙光人才计划、上海市科委青年科技英才扬帆计划等。担任中国微生物学会微生物组专业委员会委员、中国生物物理学会肠道菌群分会委员、上海市微生物学会基础微生物专业委员会委员、上海市预防医学会微生态专业委员会委员等。研究方向为营养调控肠道菌群与代谢性疾病,以第一作者或通讯作者在国际上具有影响力的杂志Science、Cell、Cell Metabolism、Nature communications、Microbiome、Gut Microbes等发表论文。2022-2024连续三年入选科睿唯安 “全球高被引科学家”。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期
iMetaOmics封面

1卷1期

1卷2期

2卷1期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,中科院分区生物学1区Top,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,目标是成为影响因子大于10的高水平综合期刊,欢迎投稿!
"iMetaMed" 是“iMeta” 子刊,专注于医学、健康和生物技术领域,目标是成为影响因子大于15的医学综合类期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
出版社iMetaMed主页:
https://onlinelibrary.wiley.com/journal/3066988x
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
iMetaMed投稿:
https://wiley.atyponrex.com/submission/dashboard?siteName=IMM3
邮箱:
office@imeta.science

智能硬件社区聚焦AI智能硬件技术生态,汇聚嵌入式AI、物联网硬件开发者,打造交流分享平台,同步全国赛事资讯、开展 OPC 核心人才招募,助力技术落地与开发者成长。
更多推荐



 0
0 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)